Ab 5 Mai 2017
Am Freitag, den 5. Mai 2017 sind die neuen EU-Verordnungen MDR (2017/745) und IVDR (2017/746) in der „Official Journal of the European Union“ veröffentlicht worden und sind ab dem 26. Mai 2017 verpflichtend für die Medizintechnik-Unternehmen, die in der Europäischen Union ihre Medizingeräte und -produkte vermarkten wollen.
Sowohl für große als auch für kleine Medizintechnik-Unternehmen ist es ratsam, die finalen Texte zu lesen und mit den Vorbereitungen der Übergangsprozesse so schnell wie möglich zu beginnen.
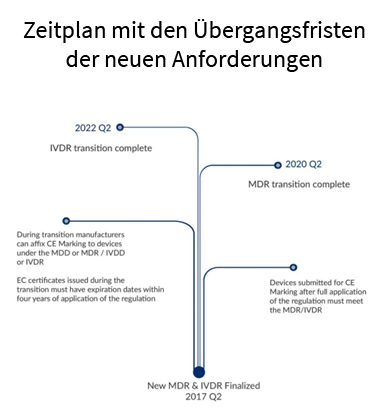
Mit der Implementierung werden die Richtlinien für Medizinprodukte (93/42/EWG) und aktive implantierbare Medizinprodukte (90/385/EWG) durch die Medical Device Regulation (MDR) zusammengeführt und abgelöst. Nach einer Übergangsfrist von drei Jahren verlieren diese Richtlinien ihre Gültigkeit. Ab 25. Mai 2020 ist dann die MDR allein gültig. Die Richtlinie für In-vitro-Diagnostika (98/79/EG) verliert nach einer Übergangsfrist von fünf Jahren ihre Gültigkeit. Ab 25. Mai 2022 ist damit die IVDR allein gültig.
Allerdings ist in Abhängigkeit der gewählten Route weiterhin möglich, ein Medizinprodukt bis 26. Mai 2024 nach der o.g. Richtlinie im europäischen Markt und somit nach dem Medizinproduktegesetz in Deutschland und nach den nationalen Gesetzen in der EU und EWR einzuführen. Diese Möglichkeit ist aber ein spezielles Szenario. Die Mehrheit der Produkte werden erwartungsgemäß nicht später als fünf Jahre ab dem 26. Mai 2017 re-zertifiziert sein.
Weitere Übergangsfristen sind in Artikel 120 (MDR) und Artikel 110 (IVDR) zu finden. Außerdem ist es wichtig zu wissen, dass es beim Geltungsbeginn der Verordnungen eine ausreichende Zahl von Benannten Stellen (Notified Bodies) gemäß den neuen Bestimmungen geben wird, damit Marktengpässe, wie sie aktuell bestehen, vermieden werden.
Nach Ansicht des Medizintechnik-Verbandes BV-Med ist die Übergangsfrist für die neue EU-Medizinprodukteverordnung angesichts der umfangreichen neuen Anforderungen an die Benannten Stellen, an die klinische Bewertung von Medizinprodukten, durch neue Pläne- und Berichtspflichten und das neue Eudamed-/UDI-Datenbanksystem allerdings knapp bemessen. Vorerst gilt noch die nationalen Medizinproduktegesetze und die entsprechenden EU-Richtlinien MDD, AIMDD und IVDD. Ratsam ist aber, dass der Hersteller sich schon heute auf die neuen bzw. weiterentwickelten Anforderungen vorbereitet und mit der Umsetzung schon in diesem Jahr beginnt. Auf die nationalen Gesundheitsministerien kommt die Aufgabe hinzu, das nationale Medizinproduktegesetz (MPG) und seine Verordnungen an das neue Recht anzupassen, denn gleichlautendes oder widersprüchliches nationales Recht ist nicht zulässig.
Es empfiehlt sich eine umfassende Analyse des Status Quo hinsichtlich
• Klassifizierung von Produkten
• Gültigkeitsdauer von Zertifikaten
• technischer Dokumentation
• bestehender Instrumente der Produktüberwachung
über den gesamten Produktlebenszyklus hinweg. Auf dieser Basis lässt sich am besten identifizieren, welche weiteren Anstrengungen unternommen werden müssen, um Unternehmen in Übereinstimmung mit der MDR bzw. IVDR aufzustellen und eine CE-Kennzeichnung nach der neuen MDR / IVDR vornehmen zu können.
Insbesondere kleine und mittelständische Unternehmen werden Wege finden müssen, die wesentlich höheren Anforderungen an Qualitäts- und Risikomanagement, technische Dokumentation und die Erstellung klinischer Daten beziehungsweise der klinischen Bewertung zu erfüllen und hierbei gleichzeitig auch den Anforderungen der UDI-Kennzeichnung und gesteigerten Reportpflichten zu genügen. So fordert die MDR periodische, teilweise jährliche Updates von Post-Market-Surveillance-Plans/Reports, Post Market Clinical, Periodic-Safety-Update-Report und Summary of Safety and Clinical Performance.
orangeglobal -
Ihr Berater für die Konkretisierung Ihrer Planung bis zur Marktüberwachung Ihres Medizinproduktes im lokalen Markt
DAS ORANGEGLOBAL LEISTUNGSPAKET IM DETAIL:
DIE AUFGABE:
• Durchführung der gesamten Palette von Dienstleistungen durch Fachexperten – ob Teilaufgaben oder
komplette Projekte.
• Ermöglichung des internationalen Marktzugangs.
• Erstellung einer zeit- und kosteneffizienten Strategie für die praxisrelevante Projektdurchführung.
DER LÖSUNGSPROZESS:
• Machbarkeits- und Bedarfsanalyse (Machbarkeitsstudie) inkl. Beratung und Klärung der Anforderungen.
• Internationale Marktforschung und Strategiekonzepte sowie Kampagnenentwicklung und -adaption durch das
Centre of Competence Marketing.
• Klinische Prüfungen mit einem klinischen Prüfplan nach ICH-GCP, ISO 14155 und MEDDEV 2.7.1 Rev. 4 für
einen reibungslosen Behördengang.
• Erstellung des Qualitätsmanagementsystems nach ISO 13485 und GMP.
• Übernahme der technischen Dokumentation gemäß den Anforderungen der MEDDEV und ZLG.
• Berücksichtigung der Nutzungsbewertung im Verlauf des Pre-Market-Testing.
• Erstellung und Übersetzung der „Instruction for Use (IFU)“ sowie der Lesbarkeitsprüfung und des Labelling
eines Medizinproduktes.
• Erstellung von eIFUs gemäß der Verordnung (EU) Nr. 207/2012.
• Usability-Testing nach Anhang I der Verordnungen MDR/IVDR
• Planung und Prüfung der Gebrauchstauglichkeit / Verwendbarkeit von Medizinprodukten nach
der Norm IEC 62366.
• Risikoanalyse und Risikobewertung zum Nachweis der Sicherheit nach DIN EN 62366 und ISO 14971.
• CE-Kennzeichnung, Notifizierungen, Registrierungen und Genehmigungen nach MDR und IVDR oder
MDD/AIMDD/IVDD.
• Kommunikation mit von Ihnen gewünschten Benannten Stellen
• Meldungen an die Behörden während der Postmarketing Surveillance.
IHR NUTZEN:
• Zugang zu lokalen Märkten rund um den Globus.
• Qualitätssteigerung und erhöhte Sicherheit von Produkt, Dokumentation und Prozessen.
• Kostensenkung durch weniger Arbeits- und Administrationsaufwand und mehr Koordinationseffizienz.
• Verkürzte internationale Time-to-Market.TUNGSPAKET
INTERNATIONALISIERUNG VON MEDIZINPRODUKTEN
ANSPRECHPARTNER:
Frau Dipl. Biol. Jana Wolkow
Project Manager Regulatory Affairs
Tel. +49 (0) 731 954 95 - 511
jana.wolkow@orangeglobal.(.)de